

Introduction
The classic method used to generate antibodies for research purposes is to immunize an animal with the protein of interest (or a part thereof), and allow the animal to elicit an immune response to generate antibodies that specifically bind to the immunised protein. The antibodies can then be purified from the serum or plasma. Standard protocols for this process have existed for many decades. Owing to their high specificity, such immune reagents are generally preferred over non-immune reagents.
However, antibodies generated this way have not always been used successfully or were not robust enough (1, 2) and the involvement of animals means variations in antibody performance from one animal to the next are inherent (3). Certain types of molecules, and even certain proteins, are not fit to elicit an immune response, and therefore this classical way of generating specific reagents is not always feasible. Hence the interest in synthetic (immuno-like) reagents.
When antibodies are generated and produced without the involvement of animals, they are regarded as synthetic antibodies, or recombinant antibodies (4). In addition, non-antibody based alternatives have also emerged, such as several forms of scaffold proteins displaying diversified amino acid sequences with specific affinity binding properties, chemically stabilised nucleic acid oligomers called aptamers, and molecularly imprinted polymers (MIPs). Moreover, they all have in common, a binding site optimized for highly specific and strong interaction with a particular part of the target molecule, called the epitope. This 2-part series will discuss these different groups of reagents in depth.
Part 1 focuses on synthetic immune reagents as alternatives to antibodies raised in animals. In this part we focus on recombinant antibodies and on Molecularly Imprinted Polymers (MIPs built around the analyte of choice). After, we will focus on scaffold proteins and aptamers.
Recombinant antibodies
What are they?
Unlike monoclonal antibodies produced from a hybridoma (i.e. a fusion between an antibody-producing B-cell and a myeloma cell line), the recombinant antibody is immortalised from the genetic sequence encoding the part of the immunoglobulin that determines binding to the target protein (the antigen binding site). These sequence sets can be cloned into an imunoglobulin G (IgG) expression vector, and after transfection into a cell line (for example HEK293, or Chinese Hamster Ovary-derived CHO), the production of the IgG with that specific antigen binding site by this cell line is achieved. The known DNA sequences guarantee that the clone can never be lost as these sequences can be resynthesised indefinely and recloned back into the IgG expression vector whenever necessary. The potential risk of loss of the original clone by genetic drift, hybridoma instability, etc., associated with monoclonal antibodies, is avoided.
DNA sequences of the antigen binding site
The antigen binding site is defined by the variable regions (also known as the Complimentarity Determining Regions or CDRs) of the antibody. They are built from parts of the heavy chain (VH) and the light chain (VL), located at the tip of each arm of the immunoglobulin. The smallest part of the antibody with the complete antigen binding site would be the Variable Fragment (Fv), existing of only the VH and VL regions. Such Fv has to be stabilised either by linking the two regions by a soluble and flexible peptide (creating a single chain Fv, scFv), or by involving the constant domains CL1 and CH1 to create an antigen binding fragment (Fab). The DNA sequence of the scFv or Fab is used as the defining identity of a recombinant antibody (5).
Recombinant antibody formats
Either the scFv or the Fab can be attached to the constant fragment (Fc) of choice through genetic reconstruction to recreate the complete Immunoglobulin. Smaller formats are in use as well: for example the minibody comprising the scFv attached to the CH3 domains, or the scFab that allows greater stability compared to the Fab (Fig 1).
Other variants and even smaller versions can also be generated. This include domain antibodies (dAbs, also known as single-domain antibodies or nanobodies) that merely comprise either the VH or VL regions alone (thus halving the normal set of 2 times 3 CDRs per chain). The heavy chain CDRs in the camelid VHH dAb has evolved to such an extent that it can bind small molecules like haptens (6) and large proteins (7), while also inhibiting enzymatic activity (8) by penetrating the enzyme active site (9, 10). Thus, dAbs offer great potential for pharmaceutical interventions (11).
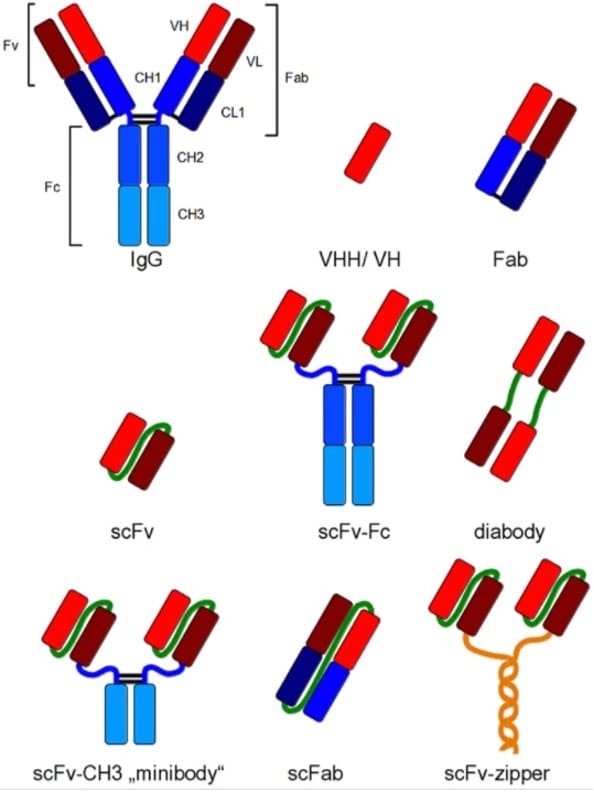
Figure 1 (Figure 1 from Frenzel et al, ref 5). Recombinant antibody formats for different applications compared to IgG. Red and dark red: variable regions; blue: constant regions; green: artificial peptide linkers; yellow: dHLX represents amphiphatic helices used for dimerization of scFv fragments.
Generation from libraries
Antigen binding sequences can be determined from an existing monoclonal antibody producing hybridoma. This, the pre-existing clone to be upgraded to an immortal recombinant antibody, enabling transfer the variable region from a mouse antibody into the IgG of a different species (for example the creation of a humanised antibody), or to change the immunoglobulin subclass.
However, most recombinant antibodies are currently generated by selection from a display library that contains large variations of scFc or Fab (or dAb) sequences. This large repertoire is obtained by PCR with VL and VH-specific primers to amplify the variable regions from the RNA of pooled B-cells (for example lymphocytes, splenocytes or peripheral blood mononuclear cells from immunized or non-immunized animals).
A naïve (or optimised to become a semisynthetic) library typically has 10 billion different sequences (12) and can be used for the selection of binding molecules to any antigen. However, a library can also be generated from immunised animals (immunised library). This could reduce the required number of variations in the library but also reducing its use to a single target (12). Library generation must be quality controlled by checking the actual number of variations and to confirmation of the correct inserts to the expression vector (Open Reading Frames, no stop codons, determining the effective size). In addition, a test panning is required to confirm that high affinity binders can be identified (4).
Antibody library validation
The quality of the identified clones depends no only on the quality of the library, but also on the quality of the screen. The correct material (high quality target and controls) against which to screen is equally important (2). In addition to screening for high affinity to the target, it is also important to counter-screen for proteins against which cross-reactivity is undesirable. When enough binders have been identified, the DNA sequence of each one is determined to identify unique candidates.
The first generation of display libraries were developed by cloning the (PCR-propagated) variable regions into a genetic contruct with a phage coat protein. After expression, using a helper phage, the result was the synthesis of phage particles with the expressed scFc, Fab or dAb displayed on their surfaces (4, 13). Subsequent panning, by selecting phage particles with the strongest binding to the target protein and reinfecting E.coli, resulted in monoclonal phages allowing sequence analysis of the selected variable domain and subsequent cloning into an IgG expression vector for recombinant antibody production by a cell line. These recombinant antibodies allow recloning of the scFv into human IgG for therapeutic applications. In addition, nanobodies (camelid VHH) are sought after for their small size.
Alternative display platform
Subsequently, alternative display platforms have been developed such as ribosome (cell-free), bacteria, yeast and mammalian display methods. Each display has advantages and disadvantages over the others. For example, the yeast display produces full-size and glycosylated IgG antibodies with very high affinity (12). This is argued to be especially useful for antibody therapies when efficacy depends on the interplay between epitope, affinity, Fc isotype and IgG glycosylation, but it has to be said that an antibody panned and selected by phage display will be naturally glycosylated by the expressing eukaryotic cell line during production anyway.
The cell-free ribosome display has the advantage of avoiding transformation or constraints imposed by the host cell (14). Although increasingly therapeutic antibodies are developed using these alternatives (12), most of the successful recombinant antibodies developed by display technology have been generated using the traditional phage display method (15). The majority of synthetic therapeutic antibodies are still derived from chimeric antibodies, i.e. when the original hybridoma-derived antibody is humanized by subcloning the variable regions into the human IgG expression vector (15).
Molecularly Imprinted Polymers
The structure of the protein epitope is mirrored by the structure of the antibody’s variable regions. Other specific binders follow the same principle. In general, strong, specific binders are selected from a large set of candidates through the panning process to identify the binders with the best fit to the target.
However, when it comes to Molecularly Imprinted Polymers (MIPs), the principle is the inverse; We start with the taget (print molecule), and subsequently build the matching binding site around it, piece by piece, through polymerising functional monomers capable of forming hydrogen bonds with the different parts of the print molecule. A cross-linker is used to facilitate the polymerisation reaction. Thus, a perfectly fitting mold is built around the print molecule, a rigid insoluble polymer, complementary to the print molecule in both shape and chemical functionality. After removal of the print molecule, the imprinted polymer is available to bind the target again, see figure 2.
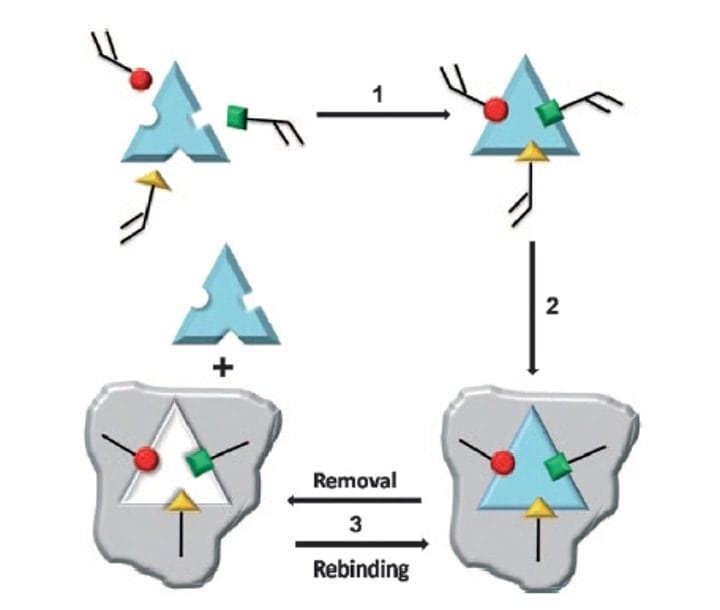
Figure 2 (Scheme 1 from Ge and Turner, ref 16). Step 1: Functional monomers (red, green and yellow) are mixed with the target/template molecule allowing interactions between the monomers and template (through non-covalent, covalent and/or metal-mediated interactions). Step2: A cross-linking agent is added to allow polymerisation between the template-bound monomers, to create the MIP (grey). Step 3: The template is removed from the MIP by extraction or chemical cleavage, thus making the MIP available for new target binding.
MIPS vs Antibodies
While antibodies are better suited to specifically bind larger molecules such as proteins, they have been used to detect smaller steroid-like molecules as well. However, MIPs are much better tools for the detection of smaller molecules. Drugs, like theophilline and diazepam, are equally well detected in serum by MIPs as they were by commercial enzyme-multiplied immunoassay technique (EMIT, 17). Because of the lower production costs, their stability and their reusability (16), MIPs are ideal for this pseudo-immunoassay approach when small molecules need detection and quantification.
Since its inception for the detection of theophilline and diazepam (17), MIPs have become popular. Early versions have since been improved by the introduction of polymeric beads with a much narrower size distribution (18), and by the production of beads at nanoscale (19, 20). These improvements benefit radio-labelled MIPs by simple bead preparation, high binding capacity (multiple binding sites per bead), enhanced selectivity, low cross-reactivity and excellent dispersion and suspension in solution (16). Subsequently, fluorescently labelled MIPs (22) and enzyme-linked MIPs have also been developed (23, 24), providing alternatives to traditional immunoassays. Recent developments have also led to the ability of MIPs to detect proteins (25).
MIPs are now being incorporated into new generation assays and sensing devices:
Assays
Quantum Dot (QD): CdSe/ZnS quantum dots are introduced as bright, photobleaching-tolerant fluorescent materials (26) for incorporation during the synthesis of the MIPs to selectively detect caffeine, uric acid and estriol (27), or dopamine (28).
Nanoparticles (NP): NP-MIPs have been developed for colorimetric assays for the detection of the insecticide Cartap in tea using silver NPs (29) and of the herbicide atrazine in apple juice using gold NPs (30). Current improvements are made by using NP-MIPs in lateral flow, so as to reduce the waste of chemicals involved (21).
Displacement Assays: The same principle as applies in competitive immune assays where a labelled analyte is displaced by the unlabelled analyte in the sample can be applied ot the use of MIPs. This type of assay is is still in its research stages due to insufficient sensitivity and the lack of homogenous batches of MIPs (batch-to-batch variations!), which are preventing commercialisation. However, development continues.
Biosensors
Sensing devices do not make use of standard detection labels such as colorimetric, fluorescence or radioactive. Instead, biosensor readout technologies act as transducers translating binding into a measurable signal. The incorporation of MIPs into such devices have been (partially) successful in some cases:
Electrochemical readout
Electrochemical detection through monitoring the conductivity of the MIP receptor layer (with and without binding of the target analyte) has yielded a limited number of successes (31). Furthermore, electrochemical Impedance Spectrometry (EIS) is a more advanced transducer mechanism in MIP-based sensing (32). They are also use to successfully measure endogenous histamine concentrations in intestinal fluid (33). Moreover, using arrays of carbon-nanotube tips with an imprinted non-conducting polymer coating was successful in the detection of sub-picograms/litre human ferritin and HPV-E7 protein (34). Recent reports on Point of Care (PoC) diagnostics using electrochemical MIPs claimed successful detection of ampicillin in milk (35), 17-beta estradiol in eel serum (36), and insulin in human plasma (37).
Acoustic wave sensors
The use of Quartz Crystal Microbalance (QCM)-based sensors have recently become intensely popular. The application is based on a piezoelectrical quartz crystal, sandwiched between two electrodes, which vibrate at resonance frequency in an alernating current field. MIPs coated on the crystal will influence the resonance frequency by mass shift when its target binds (MIP-QCM sensor). This method allows the detection of macromolecular targets such as proteins, viruses, bacteria and whole cells (38, 39, 40, 41, 42, 43, 44).
Thermal readout
Based on the principle of a temperature shift in the matrix when one of its components (the analyte) is caught by the stationary phase (in this case the MIP layer), the combination of two thermocouples (one at the base and one inside the matrix) will measure the thermal wave. The measured shift will be proportional to the quantity of bound analyte. Thus, the concentration of dopamine in serum and urine could be established in the 10-60uM range (45).
Optical sensors
MIPs are involved with both Surface Plasmon Resonance (SPR) and Surface-Enhanced Raman Spectrometry (SERS) as optical transducer. In the SPR-MIP sensor, the binding of the analyte to the MIP will cause a change of electron behaviour resulting in a shift in the light reflection at the surface of the metal MIP carrier. This shift is proportional to the concentration of analyte above the MIP layer, thus the detection of viruses (46), dansylated amino acids (47), lysozyme (48), antibiotics (49) anti-cancer drug (50) and cardiac biomarker (51).
In SERS, the so-called Raman scattering of light will respond to the degree of binding of the analyte to the binding surface (in this case the MIP). A recent report claimed detection of a tumour biomarker in human blood (52), but most emphasis is on food safety and environmental monitoring (21).
References
- Baker M. When antibodies mislead: the quest for validation. Nature. 2020 Sep;585(7824):313-314. doi: 10.1038/d41586-020-02549-1. PMID: 32895531.
- Voskuil JLA, Bandrowski A, Begley CG, Bradbury ARM, Chalmers AD, Gomes AV, Hardcastle T, Lund-Johansen F, Plückthun A, Roncador G, Solache A, Taussig MJ, Trimmer JS, Williams C, Goodman SL. The Antibody Society’s antibody validation webinar series. MAbs. 2020 Jan-Dec;12(1):1794421. doi: 10.1080/19420862.2020.1794421. PMID: 32748696; PMCID: PMC7531563.
- Voskuil JLA. The challenges with the validation of research antibodies. F1000Res. 2017 Feb 17;6:161. doi: 10.12688/f1000research.10851.1. PMID: 28357047; PMCID: PMC5333605.
- Miersch S, Sidhu SS. Synthetic antibodies: concepts, potential and practical considerations. Methods. 2012 Aug;57(4):486-98. doi: 10.1016/j.ymeth.2012.06.012. Epub 2012 Jun 27. PMID: 22750306
- Frenzel A, Hust M, Schirrmann T. Expression of recombinant antibodies. Front Immunol. 2013 Jul 29;4:217. doi: 10.3389/fimmu.2013.00217. PMID: 23908655; PMCID: PMC3725456.
- Spinelli S, Frenken LG, Hermans P, Verrips T, Brown K, Tegoni M, Cambillau C. Camelid heavy-chain variable domains provide efficient combining sites to haptens. Biochemistry. 2000 Feb 15;39(6):1217-22. doi: 10.1021/bi991830w. PMID: 10684599
- Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997 Sep 15;414(3):521-6. doi: 10.1016/s0014-5793(97)01062-4. PMID: 9323027.
- Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Hölzer W, De Genst E, Wyns L, Muyldermans S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998 Jul 1;17(13):3512-20. doi: 10.1093/emboj/17.13.3512. PMID: 9649422; PMCID: PMC1170688.
- Desmyter A, Transue TR, Ghahroudi MA, Thi MH, Poortmans F, Hamers R, Muyldermans S, Wyns L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol. 1996 Sep;3(9):803-11. doi: 10.1038/nsb0996-803. PMID: 8784355.
- Desmyter A, Spinelli S, Payan F, Lauwereys M, Wyns L, Muyldermans S, Cambillau C. Three camelid VHH domains in complex with porcine pancreatic alpha-amylase. Inhibition and versatility of binding topology. J Biol Chem. 2002 Jun 28;277(26):23645-50. doi: 10.1074/jbc.M202327200. Epub 2002 Apr 17. PMID: 11960990.
- Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003 Nov;21(11):484-90. doi: 10.1016/j.tibtech.2003.08.007. PMID: 14573361.
- Almagro JC, Pedraza-Escalona M, Arrieta HI, Pérez-Tapia SM. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies (Basel). 2019 Aug 23;8(3):44. doi: 10.3390/antib8030044. PMID: 31544850; PMCID: PMC6784186.
- McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990 Dec 6;348(6301):552-4. doi: 10.1038/348552a0. PMID: 2247164.
- Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A. 1997 May 13;94(10):4937-42. doi: 10.1073/pnas.94.10.4937. PMID: 9144168; PMCID: PMC24609.
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020 Jan 2;27(1):1. doi: 10.1186/s12929-019-0592-z. PMID: 31894001; PMCID: PMC6939334
- Ge Y, Turner AP. Molecularly imprinted sorbent assays: recent developments and applications. Chemistry. 2009 Aug 17;15(33):8100-7. doi: 10.1002/chem.200802401. PMID: 19630010
- Vlatakis G, Andersson LI, Müller R, Mosbach K. Drug assay using antibody mimics made by molecular imprinting. Nature. 1993 Feb 18;361(6413):645-7. doi: 10.1038/361645a0. PMID: 8437624
- Kempe H, Kempe M. Development and evaluation of spherical molecularly imprinted polymer beads. Anal Chem. 2006 Jun 1;78(11):3659-66. doi: 10.1021/ac060068i. PMID: 16737221
- Yoshimatsu K, Reimhult K, Krozer A, Mosbach K, Sode K, Ye L. Uniform molecularly imprinted microspheres and nanoparticles prepared by precipitation polymerization: the control of particle size suitable for different analytical applications. Anal Chim Acta. 2007 Feb 12;584(1):112-21. doi: 10.1016/j.aca.2006.11.004. Epub 2006 Nov 10. Erratum in: Anal Chim Acta. 2010 Jan 11;657(2):215. PMID: 17386593
- Wei S, Molinelli A, Mizaikoff B. Molecularly imprinted micro and nanospheres for the selective recognition of 17beta-estradiol. Biosens Bioelectron. 2006 Apr 15;21(10):1943-51. doi: 10.1016/j.bios.2005.09.017. Epub 2005 Dec 2. PMID: 16326090
- Lowdon JW, Diliën H, Singla P, Peeters M, Cleij TJ, van Grinsven B, Eersels K. MIPs for commercial application in low-cost sensors and assays – An overview of the current status quo. Sens Actuators B Chem. 2020 Dec 15;325:128973. doi: 10.1016/j.snb.2020.128973. Epub 2020 Sep 30. PMID: 33012991
- Turkewitsch P, Wandelt B, Darling GD, Powell WS. Fluorescent Functional Recognition Sites through Molecular Imprinting. A Polymer-Based Fluorescent Chemosensor for Aqueous cAMP. Anal Chem. 1998 Jul 1;70(13):2771. doi: 10.1021/ac981558g. PMID: 21644794
- Piletsky SA, Piletska EV, Bossi A, Karim K, Lowe P, Turner AP. Substitution of antibodies and receptors with molecularly imprinted polymers in enzyme-linked and fluorescent assays. Biosens Bioelectron. 2001 Dec;16(9-12):701-7. doi: 10.1016/s0956-5663(01)00234-2. PMID: 11679247
- Peng D, Li Z, Wang Y, Liu Z, Sheng F, Yuan Z. Enzyme-linked immunoassay based on imprinted microspheres for the detection of sulfamethazine residue. J Chromatogr A. 2017 Jul 14;1506:9-17. doi: 10.1016/j.chroma.2017.05.016. Epub 2017 May 8. PMID: 28545731
- Xu J, Medina-Rangel PX, Haupt K, Tse Sum Bui B. Guide to the Preparation of Molecularly Imprinted Polymer Nanoparticles for Protein Recognition by Solid-Phase Synthesis. Methods Enzymol. 2017;590:115-141. doi: 10.1016/bs.mie.2017.02.004. Epub 2017 Mar 17. PMID: 28411635
- Suzuki M, Udaka H, Fukuda T. Quantum dot-linked immunosorbent assay (QLISA) using orientation-directed antibodies. J Pharm Biomed Anal. 2017 Sep 5;143:110-115. doi: 10.1016/j.jpba.2017.05.014. Epub 2017 May 29. PMID: 28582666
- Lin CI, Joseph AK, Chang CK, Lee YD. Molecularly imprinted polymeric film on semiconductor nanoparticles analyte detection by quantum dot photoluminescence. J Chromatogr A. 2004 Feb 20;1027(1-2):259-62. doi: 10.1016/j.chroma.2003.10.037. PMID: 14971510
- Wang J, Dai J, Xu Y, Dai X, Zhang Y, Shi W, Sellergren B, Pan G. Molecularly Imprinted Fluorescent Test Strip for Direct, Rapid, and Visual Dopamine Detection in Tiny Amount of Biofluid. Small. 2019 Jan;15(1):e1803913. doi: 10.1002/smll.201803913. Epub 2018 Nov 23. PMID: 30468558.
- Wu M, Deng H, Fan Y, Hu Y, Guo Y, Xie L. Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements. Molecules. 2018 Jun 14;23(6):1443. doi: 10.3390/molecules23061443. PMID: 29899218
- Zhao B, Feng S, Hu Y, Wang S, Lu X. Rapid determination of atrazine in apple juice using molecularly imprinted polymers coupled with gold nanoparticles-colorimetric/SERS dual chemosensor. Food Chem. 2019 Mar 15;276:366-375. doi: 10.1016/j.foodchem.2018.10.036. Epub 2018 Oct 9. PMID: 30409607
- Yaroshenko I, Kirsanov D, Marjanovic M, Lieberzeit PA, Korostynska O, Mason A, Frau I, Legin A. Real-Time Water Quality Monitoring with Chemical Sensors. Sensors (Basel). 2020 Jun 17;20(12):3432. doi: 10.3390/s20123432. PMID: 32560552
- Chen L, Wang X, Lu W, Wu X, Li J. Molecular imprinting: perspectives and applications. Chem Soc Rev. 2016 Apr 21;45(8):2137-211. doi: 10.1039/c6cs00061d. Epub 2016 Mar 3. PMID: 26936282
- Wackers G, Putzeys T, Peeters M, Van de Cauter L, Cornelis P, Wübbenhorst M, Tack J, Troost F, Verhaert N, Doll T, Wagner P. Towards a catheter-based impedimetric sensor for the assessment of intestinal histamine levels in IBS patients. Biosens Bioelectron. 2020 Jun 15;158:112152. doi: 10.1016/j.bios.2020.112152. Epub 2020 Mar 20. PMID: 32275205
- Cai D, Ren L, Zhao H, Xu C, Zhang L, Yu Y, Wang H, Lan Y, Roberts MF, Chuang JH, Naughton MJ, Ren Z, Chiles TC. A molecular-imprint nanosensor for ultrasensitive detection of proteins. Nat Nanotechnol. 2010 Aug;5(8):597-601. doi: 10.1038/nnano.2010.114. Epub 2010 Jun 27. PMID: 20581835
- Liu Z, Fan T, Zhang Y, Ren X, Wang Y, Ma H, Wei Q. Electrochemical assay of ampicillin using Fe3N-Co2N nanoarray coated with molecularly imprinted polymer. Mikrochim Acta. 2020 Jul 13;187(8):442. doi: 10.1007/s00604-020-04432-2. PMID: 32661724
- Lee MH, Thomas JL, Su ZL, Zhang ZX, Lin CY, Huang YS, Yang CH, Lin HY. Doping of transition metal dichalcogenides in molecularly imprinted conductive polymers for the ultrasensitive determination of 17β-estradiol in eel serum. Biosens Bioelectron. 2020 Feb 15;150:111901. doi: 10.1016/j.bios.2019.111901. Epub 2019 Nov 18. PMID: 31767344
- Garcia Cruz A, Haq I, Cowen T, Di Masi S, Trivedi S, Alanazi K, Piletska E, Mujahid A, Piletsky SA. Design and fabrication of a smart sensor using in silico epitope mapping and electro-responsive imprinted polymer nanoparticles for determination of insulin levels in human plasma. Biosens Bioelectron. 2020 Dec 1;169:112536. doi: 10.1016/j.bios.2020.112536. Epub 2020 Aug 28. PMID: 32980804
- Tai DF, Lin CY, Wu TZ, Chen LK. Recognition of dengue virus protein using epitope-mediated molecularly imprinted film. Anal Chem. 2005 Aug 15;77(16):5140-3. doi: 10.1021/ac0504060. PMID: 16097751
- Lin TY, Hu CH, Chou TC. Determination of albumin concentration by MIP-QCM sensor. Biosens Bioelectron. 2004 Jul 30;20(1):75-81. doi: 10.1016/j.bios.2004.01.028. PMID: 15142579
- Tai DF, Jhang MH, Chen GY, Wang SC, Lu KH, Lee YD, Liu HT. Epitope-cavities generated by molecularly imprinted films measure the coincident response to anthrax protective antigen and its segments. Anal Chem. 2010 Mar 15;82(6):2290-3. doi: 10.1021/ac9024158. PMID: 20184289.
- Dickert FL, Hayden O, Bindeus R, Mann KJ, Blaas D, Waigmann E. Bioimprinted QCM sensors for virus detection-screening of plant sap. Anal Bioanal Chem. 2004 Apr;378(8):1929-34. doi: 10.1007/s00216-004-2521-5. Epub 2004 Feb 18. PMID: 14985911
- Poller AM, Spieker E, Lieberzeit PA, Preininger C. Surface Imprints: Advantageous Application of Ready2use Materials for Bacterial Quartz-Crystal Microbalance Sensors. ACS Appl Mater Interfaces. 2017 Jan 11;9(1):1129-1135. doi: 10.1021/acsami.6b13888. Epub 2016 Dec 21. PMID: 27936575
- Latif U, Qian J, Can S, Dickert FL. Biomimetic receptors for bioanalyte detection by quartz crystal microbalances – from molecules to cells. Sensors (Basel). 2014 Dec 5;14(12):23419-38. doi: 10.3390/s141223419. PMID: 25490598
- Jenik M, Seifner A, Krassnig S, Seidler K, Lieberzeit PA, Dickert FL, Jungbauer C. Sensors for bioanalytes by imprinting–polymers mimicking both biological receptors and the corresponding bioparticles. Biosens Bioelectron. 2009 Sep 15;25(1):9-14. doi: 10.1016/j.bios.2009.01.019. Epub 2009 Jan 23. PMID: 19231153
- Diliën H, Peeters M, Royakkers J, Harings J, Cornelis P, Wagner P, Steen Redeker E, Banks CE, Eersels K, van Grinsven B, Cleij TJ. Label-Free Detection of Small Organic Molecules by Molecularly Imprinted Polymer Functionalized Thermocouples: Toward In Vivo Applications. ACS Sens. 2017 Apr 28;2(4):583-589. doi: 10.1021/acssensors.7b00104. Epub 2017 Apr 13. PMID: 28480332
- Altintas Z, Gittens M, Guerreiro A, Thompson KA, Walker J, Piletsky S, Tothill IE. Detection of Waterborne Viruses Using High Affinity Molecularly Imprinted Polymers. Anal Chem. 2015 Jul 7;87(13):6801-7. doi: 10.1021/acs.analchem.5b00989. Epub 2015 Jun 9. PMID: 26008649
- Li X, Husson SM. Adsorption of dansylated amino acids on molecularly imprinted surfaces: a surface plasmon resonance study. Biosens Bioelectron. 2006 Sep 15;22(3):336-48. doi: 10.1016/j.bios.2006.04.016. Epub 2006 Jun 6. PMID: 16753292
- Matsunaga T, Hishiya T, Takeuchi T. Surface plasmon resonance sensor for lysozyme based on molecularly imprinted thin films. Anal Chim Acta. 2007 May 15;591(1):63-7. doi: 10.1016/j.aca.2007.02.072. Epub 2007 Mar 12. PMID: 17456425
- Altintas Z. Surface plasmon resonance based sensor for the detection of glycopeptide antibiotics in milk using rationally designed nanoMIPs. Sci Rep. 2018 Jul 25;8(1):11222. doi: 10.1038/s41598-018-29585-2. PMID: 30046057
- Özkan A, Atar N, Yola ML. Enhanced surface plasmon resonance (SPR) signals based on immobilization of core-shell nanoparticles incorporated boron nitride nanosheets: Development of molecularly imprinted SPR nanosensor for anticancer drug, etoposide. Biosens Bioelectron. 2019 Apr 1;130:293-298. doi: 10.1016/j.bios.2019.01.053. Epub 2019 Jan 31. PMID: 30776616
- Baldoneschi V, Palladino P, Banchini M, Minunni M, Scarano S. Norepinephrine as new functional monomer for molecular imprinting: An applicative study for the optical sensing of cardiac biomarkers. Biosens Bioelectron. 2020 Jun 1;157:112161. doi: 10.1016/j.bios.2020.112161. Epub 2020 Mar 20. PMID: 32250934
- Lin X, Wang Y, Wang L, Lu Y, Li J, Lu D, Zhou T, Huang Z, Huang J, Huang H, Qiu S, Chen R, Lin D, Feng S. Interference-free and high precision biosensor based on surface enhanced Raman spectroscopy integrated with surface molecularly imprinted polymer technology for tumor biomarker detection in human blood. Biosens Bioelectron. 2019 Oct 15;143:111599. doi: 10.1016/j.bios.2019.111599. Epub 2019 Aug 21. PMID: 31476600
- Yan M, She Y, Cao X, Ma J, Chen G, Hong S, Shao Y, Abd El-Aty AM, Wang M, Wang J. A molecularly imprinted polymer with integrated gold nanoparticles for surface enhanced Raman scattering based detection of the triazine herbicides, prometryn and simetryn. Mikrochim Acta. 2019 Feb 1;186(3):143. doi: 10.1007/s00604-019-3254-7. Erratum in: Mikrochim Acta. 2019 Mar 5;186(4):215. PMID: 30707371


